1. The Statutory Standard and the Evolution of Substantial Evidence
For a drug to achieve FDA approval, applicants must provide "substantial evidence of effectiveness" under Section 505 of the FD&C Act. This statutory standard requires evidence consisting of adequate and well-controlled investigations, enabling experts to responsibly conclude that the drug will have its purported effect.
It is essential to distinguish the effect of the drug "from other influences, such as spontaneous change in the course of the disease, placebo effect, or biased observation." Similarly, under section 351(a) of the Public Health Service Act, biological products must be shown to be "safe, pure, and potent," with potency long interpreted by the FDA to include effectiveness.
While the "two-trial dogma" has been the historical default, regulatory modernization has formally codified the FDA's authority to consider data from a single adequate and well-controlled clinical investigation, alongside confirmatory evidence obtained before or after the investigation, as constituting substantial evidence. This paradigm shift does not lower the evidentiary bar; instead, it reframes how substantial evidence is demonstrated by emphasizing statistical rigor, mechanistic understanding, and post-market data.
2. Deconstructing the Factors That Impact Evidence Strength
To rely on a single pivotal trial, sponsors must ensure the investigation is scientifically unassailable. The FDA assesses the strength of evidence across multiple facets of the trial:
Trial Design and Control Selection
Design elements must be carefully selected to minimize bias and allow for a valid, quantitative assessment of the drug's effect.
- Active vs. Placebo Controls: Establishing superiority to a randomized concurrent control group (whether active or placebo) generally provides strong evidence of effectiveness.
- Non-Inferiority (NI) Trials: NI designs can be appropriate when comparing against an active control that has shown a consistent, large effect in prior superiority trials. However, the strength of evidence from an NI trial varies depending on the confidence in untested assumptions and the selected NI margin.
- Externally Controlled Trials: While these have a high potential for bias, they can support effectiveness if the natural history of the disease is well defined, the external population is highly similar to the treatment group, and the estimated treatment effect is so large that it is unlikely to be fully attributable to bias.
Endpoints and Clinical Meaningfulness
Effectiveness is typically supported by a clinical endpoint, which directly measures how a patient feels, functions, or survives.
- Use of a clinical endpoint is preferred when feasible.
- Alternatively, surrogate endpoints can be used if supported by scientific evidence. Surrogate endpoints that are "reasonably likely" to predict clinical benefit support accelerated approval, whereas validated surrogate endpoints support traditional approval.
- The FDA stresses that even small effects may be clinically meaningful if the outcome involves survival or irreversible morbidity.
Trial Conduct and Analysis
Poor execution can render a trial of any design incapable of providing substantial evidence of effectiveness.
- Trials must adhere to Good Clinical Practice (GCP) standards, maintain blinding, and minimize missing data.
- The statistical analysis should produce valid estimates aligned with a prespecified primary estimand.
- While hypothesis testing often uses a one-sided significance level of 0.025, the FDA expects a more stringent prespecified level if the prior evidence for the drug's effectiveness is not strong.
3. The Confirmatory Evidence Matrix
When relying on a single pivotal trial, the FDA expects either strong confirmatory evidence from another source or a highly persuasive standalone trial.
Pathway A: Strong Confirmatory Evidence
If the single trial is not overwhelmingly persuasive on its own, it must be paired with strong confirmatory evidence, which can include:
- Related Clinical Data: Trial data from a related indication, such as a different stage of the same disease (e.g., initial treatment vs. a treatment-refractory form of cancer) or a related disease with similar underlying pathophysiology.
- Pharmacological Class Data: Evidence from other approved drugs in the same class, based on the similarity of mechanism of action, measured endpoints, and consistency of effects across the class.
- Mechanistic and Natural History Data: Evidence showing that a drug directly targets the major driver of a disease (e.g., by correcting an enzymatic defect) can serve as confirmatory evidence. Natural history data or real-world data (RWD) can also provide reassurance that outcomes in the trial's control group accurately reflect what would occur without treatment.
Pathway B: The Highly Persuasive Trial
In circumstances where strong confirmatory evidence is unavailable, the single trial itself must be highly persuasive. The FDA expects such a trial to:
- Use a clinically meaningful primary endpoint (e.g., mortality) and have sufficient power to convincingly demonstrate an effect.
- Generate primary analysis results that are both clinically and statistically persuasive at a high level, accounting for the pretrial probability that the drug is effective.
- Generate supportive results across prespecified secondary endpoints and important trial subsets (e.g., age, sex, concomitant therapies).
- Demonstrate robustness to plausible violations in statistical assumptions.
Ethical Considerations: The FDA notes that conducting a second trial after a highly persuasive, strongly positive trial has demonstrated a substantial decrease in mortality would generally present significant ethical concerns.
4. Regulatory Flexibility and the Clinical Context
The FDA exercises regulatory flexibility in applying the substantial evidence standard, taking into account the clinical context and balancing the risk of uncertainty against the risk of delaying an effective therapy.
- Disease Severity: Flexibility is frequently applied for "life-threatening" diseases (where the likelihood of death is high) or "severely debilitating" diseases (which cause major irreversible morbidity).
- Disease Rarity: A rare disease affects fewer than 200,000 persons in the United States. Because these trials often have smaller sample sizes, the FDA emphasizes optimizing trial design with features such as covariate adjustment and appropriate endpoint selection.
- Statistical Flexibility: In certain clinical circumstances, the FDA may accept a significance level higher than the common one-sided 0.025 level, provided it is prespecified, justified, and agreed upon with the Agency.
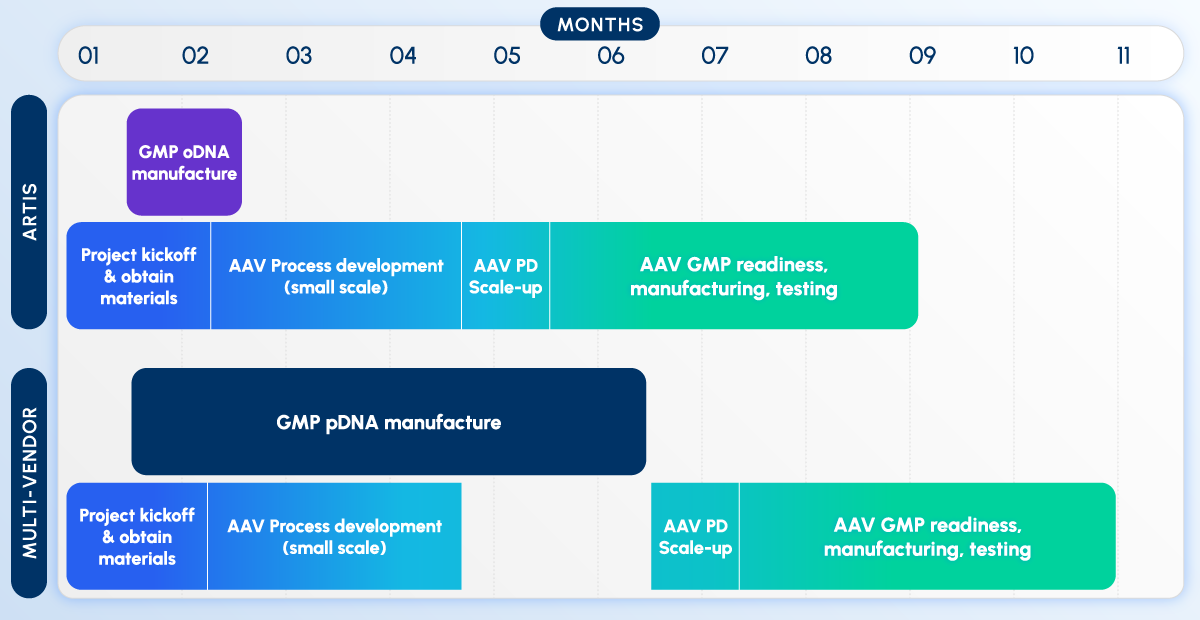
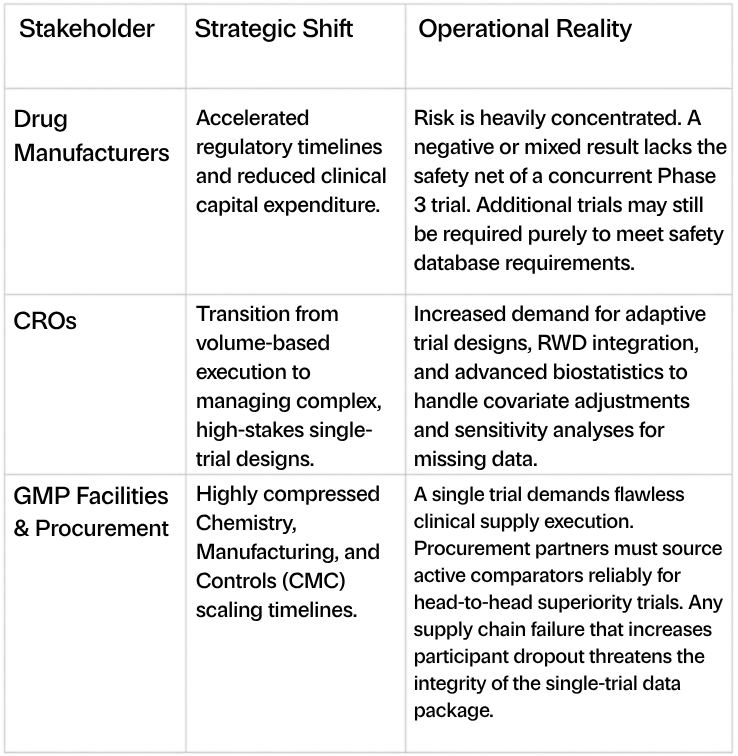
5. Implications for the Biopharma Industry: Manufacturing, CROs, Procurement, and Safety
The reliance on a single pivotal trial inherently concentrates clinical and financial risk, demanding unprecedented operational excellence across the biopharmaceutical supply chain.
The Safety Challenge
A critical caveat to the single-trial approach is that finding substantial evidence of effectiveness is necessary but not sufficient for FDA approval; the drug must also be safe. In many cases, an adequate, well-controlled trial sufficient to demonstrate efficacy may not have sufficient participant volume or treatment duration to appropriately assess safety. Therefore, sponsors may still be required to conduct a larger or longer supplementary trial solely to ensure an adequate safety database.
Stakeholder Impact
6. Case Studies in the Modern Regulatory Landscape
The reality of the FDA's evolving standards is best understood through the recent regulatory journeys of companies navigating single-trial data packages.
Outlook Therapeutics (OTLK): A Masterclass in Confirmatory Evidence
Outlook Therapeutics’ journey with ONS-5010/LYTENAVA (bevacizumab-vikg) for neovascular age-related macular degeneration (nAMD) perfectly illustrates how the FDA evaluates the "one trial plus confirmatory evidence" standard. Following a Complete Response Letter (CRL), OTLK utilized the Formal Dispute Resolution (FDR) process.
The FDA concluded that OTLK had met the substantial evidence standard by pairing a single primary trial (NORSE TWO) with an interlocking matrix of confirmatory evidence. This supplementary data included a secondary trial (NORSE EIGHT), established natural history, and robust mechanistic and pharmacodynamic data. Consequently, the FDA granted a Class 1 BLA resubmission, demonstrating that a single trial, fortified by multifaceted biological and historical evidence, is a viable pathway to approval.
Aldeyra Therapeutics (ALDX): The Risks of Inconsistent Multiple-Trial Data
Conversely, Aldeyra Therapeutics’ experience with reproxalap for dry eye disease highlights why the FDA is shifting away from simply requiring more trials and toward quality and consistency. In March 2026, ALDX received a CRL stating that the application failed to demonstrate efficacy in adequate and well-controlled studies. Crucially, the FDA noted that the "inconsistency of study results raises serious concerns about the reliability and meaningfulness of the positive findings." Because ALDX submitted a multi-trial package containing both successful and failed studies, the totality of the evidence was compromised. This case underscores a vital reality of the new regulatory paradigm: running multiple trials can actually introduce fatal inconsistency into an application, illustrating why a single, highly persuasive, and flawlessly executed pivotal trial may be a safer strategic bet for developers.
7. Conclusion
The FDA’s mandate to clarify and expand the single pivotal trial pathway represents a vital modernization of drug regulation. By integrating rigorous statistical analysis with mechanistic understanding and real-world confirmatory evidence, developers can accelerate patient access to critical therapies. However, this streamlined path concentrates risk. Success requires manufacturers, CROs, and GMP facilities to operate with unprecedented precision, utilizing elite clinical procurement partners to ensure trial integrity and navigating complex safety database requirements to ultimately secure regulatory approval.
In short, navigating this compressed, high-stakes regulatory environment requires immediate access to highly specialized external expertise. Scientist.com directly mitigates the concentrated financial and operational risks of the single-trial pathway by streamlining the sourcing and vetting of elite CROs, GMP manufacturing facilities, and real-world data (RWD) providers. Through our R&D procurement orchestration platform, biopharma developers can instantly connect with pre-vetted suppliers capable of executing complex adaptive trial designs, managing sophisticated biostatistics, and ensuring flawless clinical supply chains. Connect with Scientist.com today to build the precise supplier ecosystem your single trial demands.
Take the Next Step
Explore Suppliers
Browse trusted partners with relevant expertise
Review Capabilities
Compare services, experience, and past work
Start Your Project
Connect and begin collaborating



.png)